
过渡金属催化烯烃的位置变换是一种原子经济性转变,在天然和工业产品合成中有着广泛的应用。在过去十年中,若干相关报道通过高效贵金属(例如Ru、Rh、Pd和Ir)和非贵金属(例如Fe、Co和Ni)催化剂推动末端烯烃向内部烯烃的位置变换在一定程度上阐明了这种转化的重要性。尽管在这一领域取得了重大进展,但催化剂控制的烯烃转位选择性仍然是一个挑战。
我院汤浩教授理论计算课题组与美国印第安纳大学化学系Jeremy M. Smith教授课题组合作报道了高自旋三配位Fe(II)亚胺配合物(1-K-(18-C-6))催化的双键转位反应,通过1,3-氢质子迁移实现了1-烯烃向2-烯烃的转化。其中,双键转位是由Fe(II)中心和碱性亚胺配体的协同作用实现。DFT计算的过渡态结构揭示了烯丙基氢直接从1-烯烃转移到亚胺配体上(5TSA-B) ,随后烯丙基C1与氨基配体上的氢结合 (5TSB-1)获得相应的产物,而Fe–H物种并未参与。底物烯丙基质子的pKa决定反应的区域选择性,而且双键转位发生的转折点在于烯丙基质子的pKa ≈ 35−36处。值得一提的是:铁催化剂的高自旋(S = 2)允许反应具有良好的官能团耐受性,包括典型毒化催化剂的位点,如胺、含N-杂环、膦化物等。
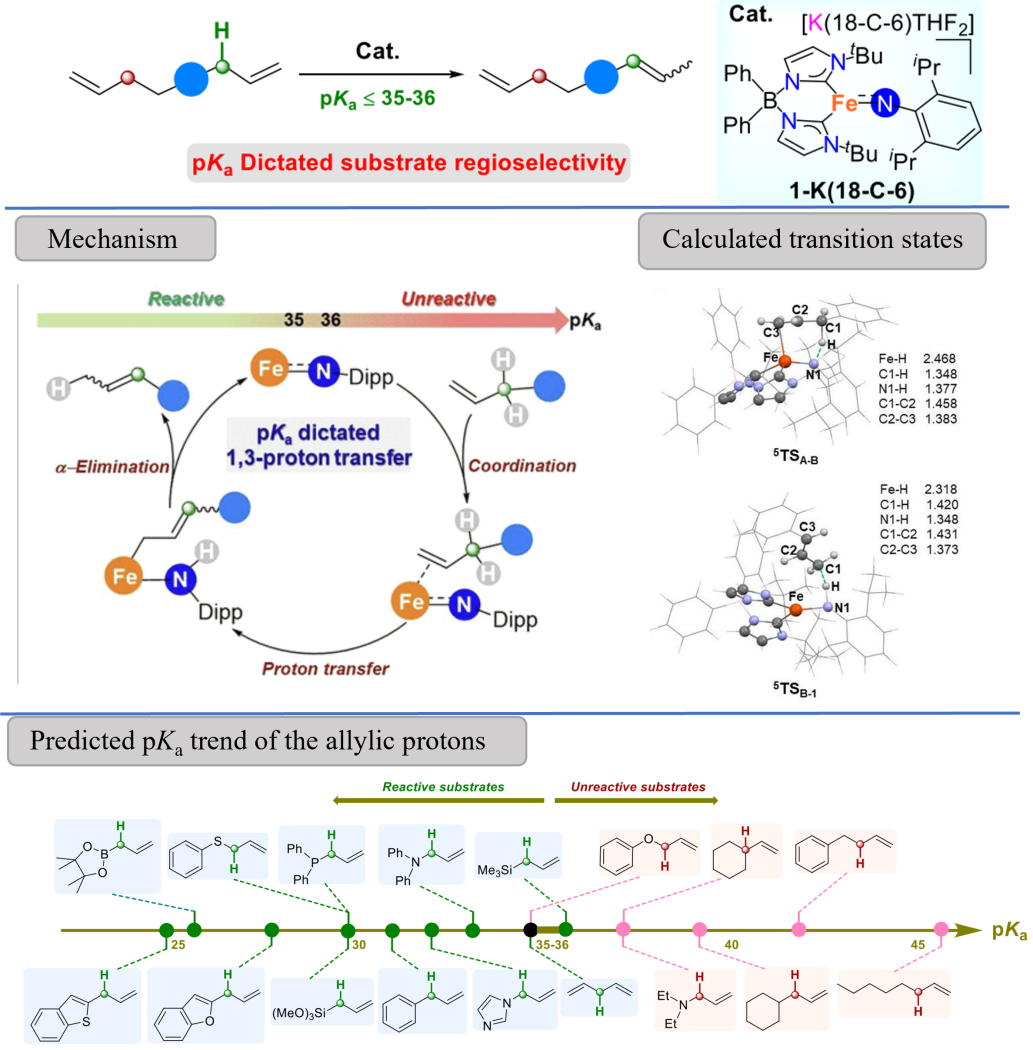
该成果以“Catalytic 1,3-Proton Transfer in Alkenes Enabled by Fe=NR Bond Cooperativity: A Strategy for pKa�Dictated Regioselective Transposition of C=C Double Bonds”为题发表在国际顶级期刊《Journal of the American Chemical Society》上。汤浩教授与美国印第安纳大学化学系Jeremy M. Smith教授为该论文共同通讯作者。
原文链接:https://pubs.acs.org/doi/10.1021/jacs.2c13350?ref=PDF
 常用链接
常用链接
 科研概况
科研概况  科研论文
科研论文