
钴基材料一直被认为是水电解中很有前途的催化剂。 特别是,许多钴基氧化物和(羟基)氢氧化物已被广泛研究作为析氧反应(OER)催化剂。 然而,由于钴基氧化物/氢氧化物对反应中间体的吸附能力强,催化活性并不理想。 目前,界面工程策略已成为调整催化活性中心金属电子结构的流行策略。该界面可能有利于富集活性位点和促进电子转移,以及优化反应中间体的吸附能,从而提高水分解效率。然而,大多数先前的报道异质结界面工作都涉及另一种化合物在基板表面上的生长,以通过表面吸附、范德华力或静电相互作用形成异质结。 这种异质结并不能够有效地增加催化活性位点,并且通过上述构筑的异质界面会由于接触界面不良而在一定程度上导致了严重的电荷交通拥堵。
近日,我院陈锡安教授课题组在纳米领域国际顶级期刊《ACS Nano》发表题为“Tuning the Interface of Co1−xS/Co(OH)F by Atomic Replacement Strategy toward High-Performance Electrocatalytic Oxygen Evolution”的论文。该工作开发了一种原子置换策略构筑高活性催化界面。典型的方法是通过简单的电沉积方法将半径较大的硫原子去置换针状结构Co(OH)F中较小的氟原子,从而形成新型异质界面。该界面具有以下优点:(1)原子取代引起的晶格畸变导致活性位点增加;(2) 通过原子置换策略在Co(OH)F表面构建的Co1-xS优化了OER过程中的吸附(OH-)和解吸(O2)能量;(3)针状结构具有尖端增强的局部电场效应。这种原子取代策略显示出对其他过渡金属硫化物(金属 = Ni、Mn、Cu)的普遍性,为构建高效电解水催化剂提供了一个新的制备策略。
为了验证半径较大的硫原子可以取代半径较小的氟原子,作者首先通过非原位XPS证明随着引入硫原子的增加氟原子相对减少;随后,通过同步辐射证明硫原子的引入能够改变Co原子配位环境的变化;最后,通过计算相应结合能的方法验证了硫原子可以取代Co(OH)F中较小的氟原子。
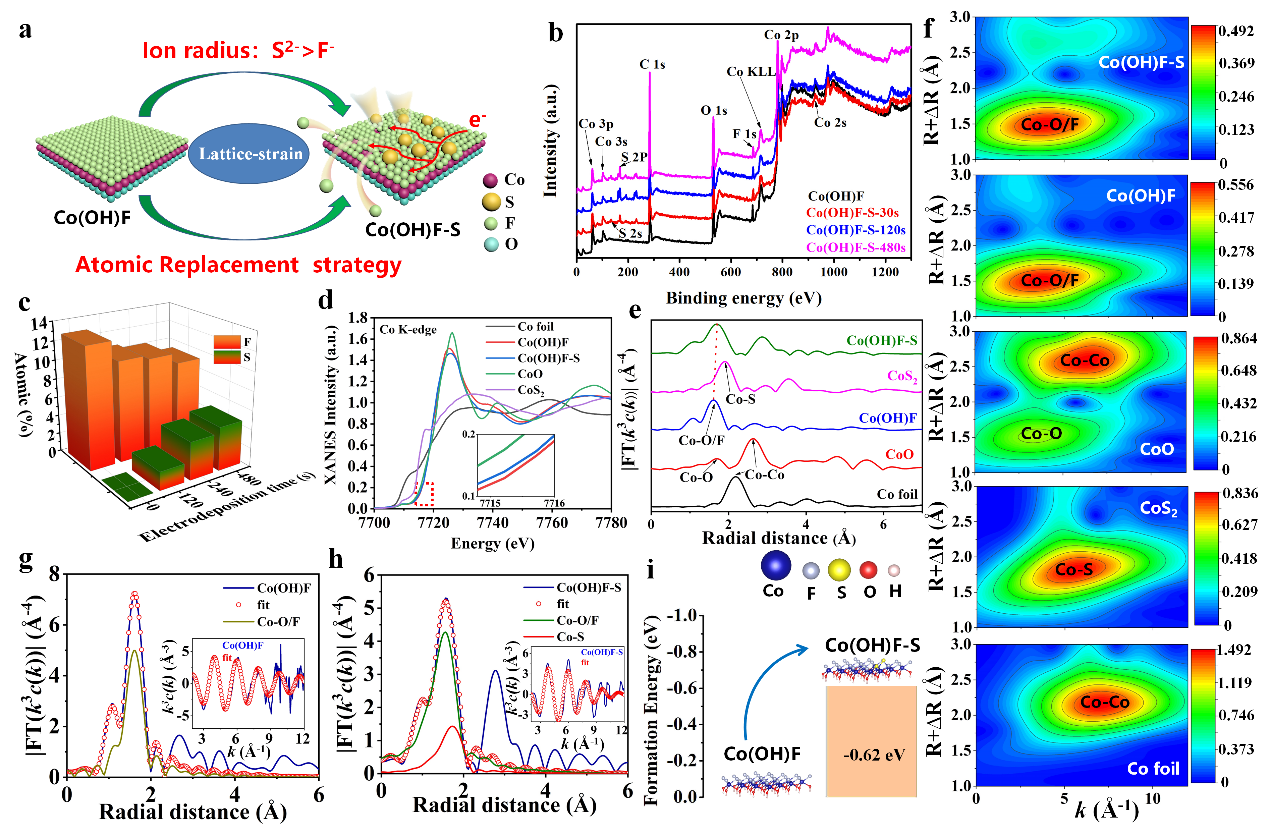
另外,通过高分辨TEM证明硫原子引入Co(OH)F中由于应力作用造成晶格畸变,这有利于催化活性位点的形成。此外,通过XPS等方法进一步证明Co1-xS通过Co-S-Co键锚定Co(OH)F上,两者之间形成强的电子相互作用。因此,Co1-xS/Co(OH)F/CC 催化剂表现出非常高的 OER 催化性能,在 10 mA cm-2 的电流密度和 71 mV dec-1 的 Tafel 斜率下过电位为 269 mV。 不对称电极在整体水分解中显示出优异的催化活性和稳定性。
相关研究结果发表于近期的《ACS Nano》(IF: 18),澳门新葡平台网址8883为第一通讯单位,学院2019级研究生林倩为第一作者,我校青年教师郭大营博士、陈锡安教授与王舜教授为该论文共同通讯作者,相关工作受到国家自然科学基金(51872208和52072273)、浙江省高层次人才特殊支持计划(2019R52042)项目的资助。
原文链接:https://pubs.acs.org/doi/pdf/10.1021/acsnano.2c07588
 常用链接
常用链接
 科研概况
科研概况  科研论文
科研论文